
The Fed’s Favorite Inflation Gauge Just Made Mortgage Relief Harder to Believe
May's PCE inflation report showed firm income, firm spending and still-hot prices, a mix that makes quick mortgage relief harder to see.
Saturday Edition
Stay updated with Archyde – your source for breaking news, global headlines, economy, entertainment, health, technology, and sports. Fresh stories daily.
May's PCE inflation report showed firm income, firm spending and still-hot prices, a mix that makes quick mortgage relief harder to see.




Continuous Coverage
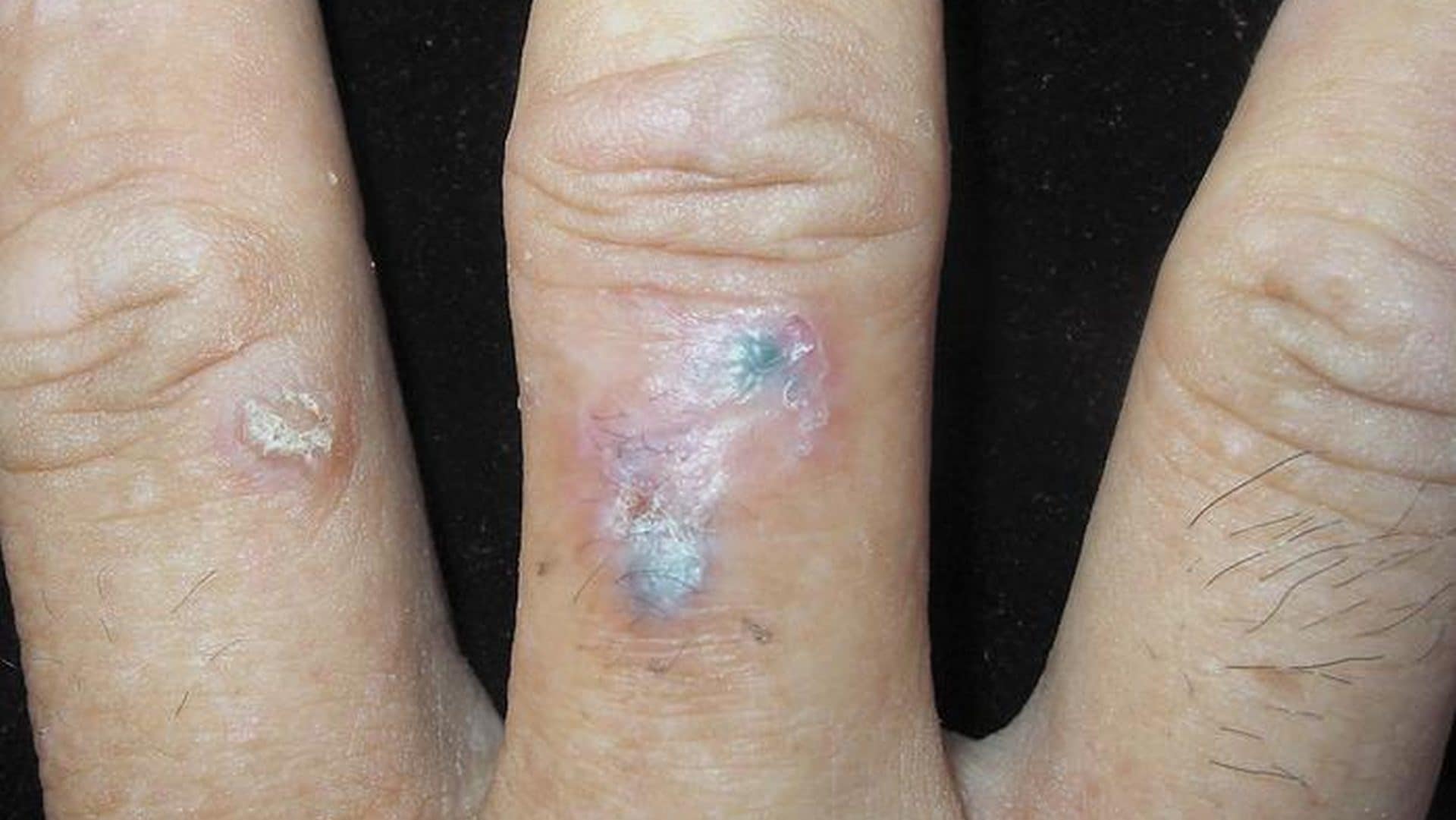
Basal cell carcinoma (BCC), a common form of skin cancer, has been identified in a rare digital presentation…
Tahkim Kurulu’ndan Mert Hakan Yandaş Kararı: Hukuki Süreçte Belirsizlik Sürüyor The Turkish Football Federation (TFF) Arbitration Board (Tahkim…

Fact-Checked Article: Global Renewable Energy Growth Surges Amid Climate Crisis In a landmark report released today, the International…

Refurbished iPhones Offer Cost-Effective Performance Refurbished iPhones, particularly the iPhone 14, provide a balance of performance, durability, and…

Players and staff from Portland San Antonio gathered for a reunion marking their 25th anniversary as Champions League…

Frontline healthcare workers battling Ebola outbreaks in the Democratic Republic of the Congo (DRC) are facing a crisis…
Global Affairs

Andy Burnham, a senior UK politician, faces calls for additional security screenings in Moscow and Makerfield, according to…
Markets And Money

Strategic Shift in the Indonesian City Car Segment Suzuki (TYO: 7269) has updated its Karimun model for 2026,…
Digital Culture

Travel influencers and digital creators are increasingly leveraging viral shopping platforms to optimize travel expenses, as seen in…
Science And Wellbeing

German racing driver Colin Jamie Bönighausen, a pilot in the ADAC GT Masters series, suffered a femoral fracture…
Screen And Sound

Twenty-five years after its 2001 theatrical debut, Disney’s Atlantis: The Lost Empire has transitioned from a box-office underperformer…
Fixtures And Form

Trick Williams Retains United States Championship Amidst Riyadh Controversy Trick Williams successfully defended his WWE United States Championship…