
{kind=link}
Breakthrough “Breathing” Chip Offers Glimpse into ALS’s Earliest Molecular Whispers
Table of Contents
- 1. Breakthrough “Breathing” Chip Offers Glimpse into ALS’s Earliest Molecular Whispers
- 2. How do miniature ALS models address the limitations of conventional animal models in ALS research?
- 3. Miniature ALS Model Offers Hope for Breakthrough Therapies
- 4. Understanding ALS and the Need for New Models
- 5. What are Miniature ALS Models?
- 6. Types of Miniature ALS Models
- 7. how Miniature ALS Models are Accelerating Research
- 8. Recent Breakthroughs and Case studies
- 9. benefits of Using Miniature ALS Models
- 10. Challenges and Future Directions
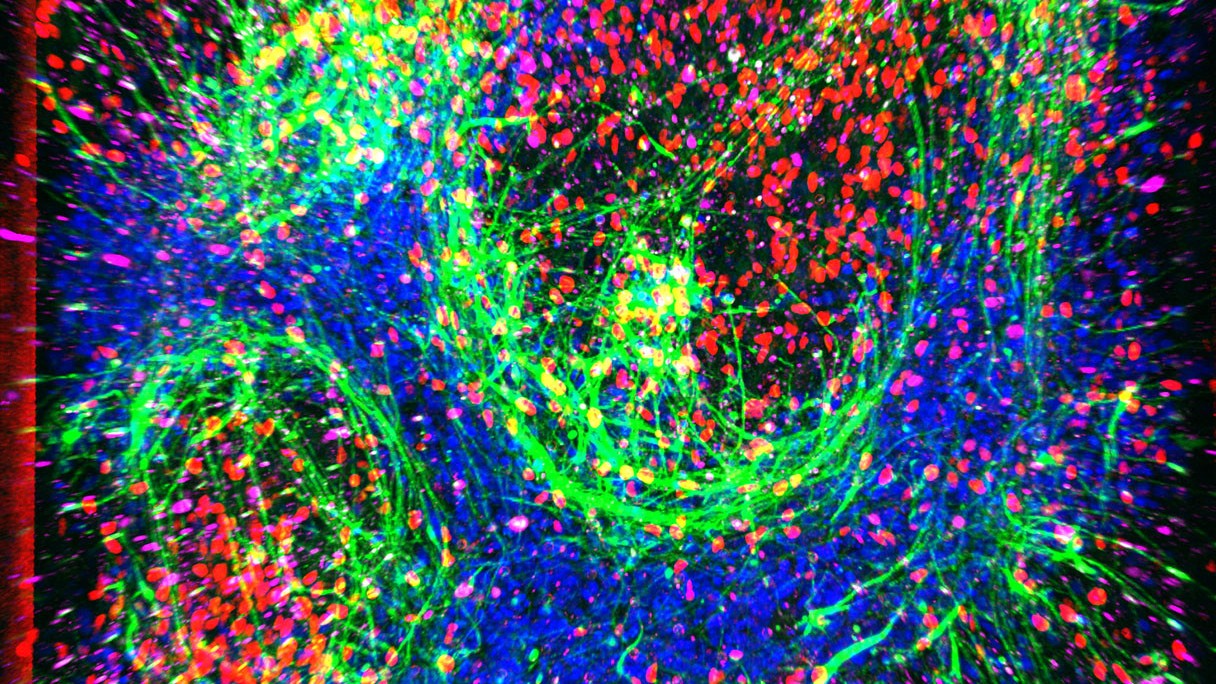
san Francisco, CA – researchers have developed a novel in vitro model that mimics the dynamic, “breathing” environment of human tissue, a notable leap forward in understanding the early molecular changes associated with Amyotrophic Lateral Sclerosis (ALS). This innovative “chip” technology is proving adept at detecting subtle differences in neurons affected by ALS, offering a powerful new tool for early diagnosis and drug growth.The advanced model, described in a recent study, recreates the complex microenvironment of human tissue, including dynamic fluid flow, which has been a critical missing element in many previous lab-based ALS studies. This feature has been lauded by self-reliant experts for its ability to enhance neuron health and maturation in the lab, allowing for the observation of early disease signals that often elude conventional models.
“Unlike most lab models that lack vascular features and dynamic flow, this chip improves neuron health and maturation,” commented Dr. Kimberly Idoko, a board-certified neurologist and medical director at Everwell Neuro, who was not involved in the research.”It captures early disease signals in ALS that are often hard to detect.”
The research team utilized their ALS and healthy control chips to conduct a complete analysis of over 10,000 genes across all the cells involved. A notably striking discovery was the identification of aberrant glutamate signaling within the neurons present in the ALS chip. Glutamate, a key excitatory neurotransmitter, typically increases the likelihood of neurons firing. In contrast, GABA is an inhibitory messenger. The study revealed heightened activity in glutamate receptor genes and diminished activity in GABA receptor genes within the motor neurons of the ALS chip when compared to the healthy control.
“We were intrigued to find this increase in glutamate activity,” stated Dr. Svendsen, a lead researcher on the project. “While we didn’t observe any visible neuron death at this stage, we hypothesize that this hyperexcitability could be a precursor to degeneration in later stages of the disease.”
This finding strongly supports long-held theories positing that excessive glutamate signaling contributes to nerve damage in ALS. Furthermore, it aligns with the mechanism of action for riluzole, a drug used to treat ALS that functions by blocking glutamate. the new chip provides compelling evidence for this mechanism and holds the potential to illuminate how it manifests in the very earliest, pre-symptomatic stages of the disease.
Dr. Idoko, while acknowledging the model’s strengths, pointed out its current limitations, noting the absence of glial cells, which play a role in ALS progression, and its inability to replicate late-stage degeneration.”However, a model like this could conceivably be useful for early drug screening, to study how a drug might cross a barrier similar to the blood-brain barrier, in readiness for animal or human studies,” she added.
The research team is actively working to extend the longevity of the cells within the model, aiming for maintenance up to 100 days. Future iterations also plan to incorporate additional cell types, such as muscle cells, to more accurately replicate the complete progression of ALS, where motor neuron loss is often accompanied by muscle wasting.
“Our goal is now to build models where more neurons die,so we can better map disease pathways and test treatments in a human-like setting,” Dr. Svendsen concluded. For the present, this advanced chip offers an unprecedented window into the earliest molecular alterations of ALS, serving as a critical tool in the quest to detect and slow the disease before irreversible damage takes hold.
How do miniature ALS models address the limitations of conventional animal models in ALS research?
Miniature ALS Model Offers Hope for Breakthrough Therapies
Understanding ALS and the Need for New Models
Amyotrophic Lateral Sclerosis (ALS), often referred to as Lou Gehrig’s disease, is a progressive neurodegenerative disease that affects nerve cells in the brain and spinal cord. This leads to muscle weakness, paralysis, and ultimately, respiratory failure. Currently, there is no cure for ALS, and treatment options are limited to managing symptoms and slowing disease progression. A significant hurdle in developing effective therapies has been the lack of accurate and accessible disease models for research. Traditional ALS models, often relying on animal studies, don’t always perfectly replicate the complexities of the human disease. This is where miniature ALS models – specifically, ALS “organoids” and “neurons-in-a-dish” – are revolutionizing the field.
What are Miniature ALS Models?
These aren’t tiny replicas of patients, but rather three-dimensional, in vitro (lab-grown) structures that mimic key aspects of the human nervous system affected by ALS. They are created using induced pluripotent stem cells (iPSCs). Here’s a breakdown:
iPSCs: These are adult cells that have been reprogrammed back into an embryonic-like state, giving them the potential to become any cell type in the body.
Differentiation: Scientists can then guide these iPSCs to differentiate into motor neurons – the nerve cells that are specifically affected in ALS.
3D Culture: These motor neurons are grown in a 3D habitat, allowing them to organize and interact with each othre in a way that more closely resembles the natural nervous system.
Patient-Specific Models: Crucially, iPSCs can be derived from skin or blood cells donated by ALS patients, creating patient-specific models that reflect the individual’s genetic makeup and disease characteristics.
Types of Miniature ALS Models
Several approaches are used to create these models, each with its strengths:
Motor Neuron Organoids: These are complex 3D structures containing various types of neural cells, including motor neurons, glial cells (which support neurons), and even vascular components. they offer a more holistic representation of the ALS microenvironment.
Neurons-in-a-Dish: These models focus on growing pure populations of motor neurons, allowing for more targeted studies of neuronal dysfunction.
Spinal Cord Organoids: Emerging research focuses on creating organoids that mimic the spinal cord, the primary site of motor neuron degeneration in ALS.
Brain Organoids: While less common for ALS specifically, brain organoids are being explored to understand the role of upper motor neurons in the disease.
how Miniature ALS Models are Accelerating Research
These models offer several advantages over traditional research methods:
- Disease modeling: They recapitulate key features of ALS pathology, such as protein aggregation (e.g., TDP-43, SOD1), neuronal loss, and impaired axonal transport.
- Drug Screening: Miniature ALS models provide a platform for high-throughput screening of potential ALS therapies. Researchers can test thousands of compounds to identify those that protect motor neurons or slow disease progression.
- Personalized Medicine: Patient-specific models allow for the testing of drugs tailored to an individual’s genetic profile,paving the way for personalized ALS treatment.
- Understanding Disease Mechanisms: By studying the cellular and molecular changes that occur in these models, scientists can gain a deeper understanding of the underlying causes of ALS.
- Reduced Reliance on Animal Models: These in vitro models can reduce the need for animal testing, addressing ethical concerns and potentially accelerating the research process.
Recent Breakthroughs and Case studies
Several promising studies have utilized miniature ALS models:
Targeting TDP-43: Researchers used motor neuron organoids to identify compounds that could reduce the aggregation of TDP-43, a protein that accumulates in the motor neurons of most ALS patients.
Gene Editing with CRISPR: Scientists employed CRISPR-Cas9 gene editing technology in patient-derived motor neurons to correct genetic mutations associated with familial ALS.
neuroinflammation Studies: Organoids have been used to investigate the role of neuroinflammation – the immune response in the brain and spinal cord – in ALS pathogenesis.
Drug Repurposing: Several existing drugs, originally developed for other conditions, are being tested in miniature ALS models for their potential to treat ALS. such as, studies are exploring the effects of certain anti-inflammatory drugs and antioxidants.
benefits of Using Miniature ALS Models
Increased Accuracy: More closely mimic human disease compared to animal models.
Cost-Effectiveness: Generally less expensive then maintaining animal colonies.
Ethical considerations: Reduces the reliance on animal testing.
Scalability: allows for high-throughput screening of potential therapies.
Personalized Approach: Enables the development of patient-specific treatments.
Challenges and Future Directions
Despite their promise, miniature ALS models are not without limitations:
Complexity: they don’t fully replicate the complexity of the entire human nervous system.
Maturation: Organoids often lack the full maturity of adult motor neurons.
Standardization: There is a need for greater standardization